Hematology

Abstract
Thrombomodulin (TM) is an endothelial anticoagulant cofactor that promotes thrombin-mediated formation of activated protein C (APC). We have found that the N-terminal lectin-like domain (D1) of TM has unique antiinflammatory properties. TM, via D1, binds high-mobility group-B1 DNA-binding protein (HMGB1), a factor closely associated with necrotic cell damage following its release from the nucleus, thereby preventing in vitro leukocyte activation, in vivo UV irradiation–induced cutaneous inflammation, and in vivo lipopolysaccharide-induced lethality. Our data also demonstrate antiinflammatory properties of a peptide spanning D1 of TM and suggest its therapeutic potential. These findings highlight a novel mechanism, i.e., sequestration of mediators, through which an endothelial cofactor, TM, suppresses inflammation quite distinctly from its anticoagulant cofactor activity, thereby preventing the interaction of these mediators with cell surface receptors on effector cells in the vasculature.
Authors
Kazuhiro Abeyama, David M. Stern, Yuji Ito, Ko-ichi Kawahara, Yasushi Yoshimoto, Motoyuki Tanaka, Tomonori Uchimura, Nobuo Ida, Yoshiaki Yamazaki, Shingo Yamada, Yasuhiko Yamamoto, Hiroshi Yamamoto, Satoshi Iino, Noboru Taniguchi, Ikuro Maruyama

Abstract
The mechanisms by which mixed-lineage leukemia (MLL) fusion products resulting from in utero translocations in 11q23 contribute to leukemogenesis and infant acute leukemia remain elusive. It is still controversial whether the MLL fusion protein is sufficient to induce acute leukemia without additional genetic alterations, although carcinogenesis in general is known to result from more than 1 genetic disorder accumulating during a lifetime. Here we demonstrate that the fusion partner–mediated homo-oligomerization of MLL-SEPT6 is essential to immortalize hematopoietic progenitors in vitro. MLL-SEPT6 induced myeloproliferative disease with long latency in mice, but not acute leukemia, implying that secondary genotoxic events are required to develop leukemia. We developed in vitro and in vivo model systems of leukemogenesis by MLL fusion proteins, where activated FMS-like receptor tyrosine kinase 3 (FLT3) together with MLL-SEPT6 not only transformed hematopoietic progenitors in vitro but also induced acute biphenotypic or myeloid leukemia with short latency in vivo. In these systems, MLL-ENL, another type of the fusion product that seems to act as a monomer, also induced the transformation in vitro and leukemogenesis in vivo in concert with activated FLT3. These findings show direct evidence for a multistep leukemogenesis mediated by MLL fusion proteins and may be applicable to development of direct MLL fusion–targeted therapy.
Authors
Ryoichi Ono, Hideaki Nakajima, Katsutoshi Ozaki, Hidetoshi Kumagai, Toshiyuki Kawashima, Tomohiko Taki, Toshio Kitamura, Yasuhide Hayashi, Tetsuya Nosaka

Abstract
Rap1b, an abundant small GTPase in platelets, becomes rapidly activated upon stimulation with agonists. Though it has been implicated to act downstream from G protein–coupled receptors (GPCRs) and upstream of integrin αIIbβ3, the precise role of Rap1b in platelet function has been elusive. Here we report the generation of a murine rap1b knockout and show that Rap1b deficiency results in a bleeding defect due to defective platelet function. Aggregation of Rap1b-null platelets is reduced in response to stimulation with both GPCR-linked and GPCR-independent agonists. Underlying the defective Rap1b-null platelet function is decreased activation of integrin αIIbβ3 in response to stimulation with agonists and signaling downstream from the integrin αIIbβ3. In vivo, Rap1b-null mice are protected from arterial thrombosis. These data provide genetic evidence that Rap1b is involved in a common pathway of integrin activation, is required for normal hemostasis in vivo, and may be a clinically relevant antithrombotic therapy target.
Authors
Magdalena Chrzanowska-Wodnicka, Susan S. Smyth, Simone M. Schoenwaelder, Thomas H. Fischer, Gilbert C. White II

Abstract
Due to its relatively slow clinical progression, B cell chronic lymphocytic leukemia (B-CLL) is classically described as a disease of accumulation rather than proliferation. However, evidence for various forms of clonal evolution suggests that B-CLL clones may be more dynamic than previously assumed. We used a nonradioactive, stable isotopic labeling method to measure B-CLL cell kinetics in vivo. Nineteen patients drank an aliquot of deuterated water (2H2O) daily for 84 days, and 2H incorporation into the deoxyribose moiety of DNA of newly divided B-CLL cells was measured by gas chromatography/mass spectrometry, during and after the labeling period. Birth rates were calculated from the kinetic profiles. Death rates were defined as the difference between calculated birth and growth rates. These analyses demonstrated that the leukemic cells of each patient had definable and often substantial birth rates, varying from 0.1% to greater than 1.0% of the entire clone per day. Those patients with birth rates greater than 0.35% per day were much more likely to exhibit active or to develop progressive disease than those with lower birth rates Thus, B-CLL is not a static disease that results simply from accumulation of long-lived lymphocytes. Rather, it is a dynamic process composed also of cells that proliferate and die, often at appreciable levels. The extent to which this turnover occurs has not been previously appreciated. A correlation between birth rates and disease activity and progression appears to exist, which may help identify patients at risk for worsening disease in advance of clinical deterioration.
Authors
Bradley T. Messmer, Davorka Messmer, Steven L. Allen, Jonathan E. Kolitz, Prasad Kudalkar, Denise Cesar, Elizabeth J. Murphy, Prasad Koduru, Manlio Ferrarini, Simona Zupo, Giovanna Cutrona, Rajendra N. Damle, Tarun Wasil, Kanti R. Rai, Marc K. Hellerstein, Nicholas Chiorazzi

Abstract
Mechanisms regulating thrombus stabilization remain largely unknown. Here, we report that loss of any 1 of the Gas6 receptors (Gas6-Rs), i.e., Tyro3, Axl, or Mer, or delivery of a soluble extracellular domain of Axl that traps Gas6 protects mice against life-threatening thrombosis. Loss of a Gas6-R does not prevent initial platelet aggregation but impairs subsequent stabilization of platelet aggregates, at least in part by reducing “outside-in” signaling and platelet granule secretion. Gas6, through its receptors, activates PI3K and Akt and stimulates tyrosine phosphorylation of the β3 integrin, thereby amplifying outside-in signaling via αIIbβ3. Blocking the Gas6-R–αIIbβ3 integrin cross-talk might be a novel approach to the reduction of thrombosis.
Authors
Anne Angelillo-Scherrer, Laurent Burnier, Nathalie Flores, Pierre Savi, Maria DeMol, Paul Schaeffer, Jean-Marc Herbert, Greg Lemke, Stephen P. Goff, Glenn K. Matsushima, H. Shelton Earp, Christian Vesin, Marc F. Hoylaerts, Stéphane Plaisance, Désiré Collen, Edward M. Conway, Bernhard Wehrle-Haller, Peter Carmeliet

Abstract
B cell chronic lymphocytic leukemia (B-CLL) is a neoplastic disorder characterized by accumulation of B lymphocytes due to uncontrolled growth and resistance to apoptosis. Analysis of B cells freshly isolated from 40 patients with chronic lymphocytic leukemia demonstrated that the Src kinase Lyn, the switch molecule that couples the B cell receptor to downstream signaling, displays anomalous properties. Lyn is remarkably overexpressed at the protein level in leukemic cells as compared with normal B lymphocytes, with a substantial aliquot of the kinase anomalously present in the cytosol. Whereas in normal B lymphocytes Lyn activation is dependent on B cell–receptor stimulation, in resting malignant cells, the constitutive activity of the kinase accounts for high basal protein tyrosine phosphorylation and low responsiveness to IgM ligation. Addition of the Lyn inhibitors PP2 and SU6656 to leukemic cell cultures restores cell apoptosis, and treatment of malignant cells with drugs that induce cell apoptosis decreases both activity and amount of the tyrosine kinase. These findings suggest a direct correlation between high basal Lyn activity and defects in the induction of apoptosis in leukemic cells. They also support a critical role for Lyn in B-CLL pathogenesis and identify this tyrosine kinase as a potential therapeutic target.
Authors
Antonella Contri, Anna Maria Brunati, Livio Trentin, Anna Cabrelle, Marta Miorin, Luca Cesaro, Lorenzo A. Pinna, Renato Zambello, Gianpietro Semenzato, Arianna Donella-Deana

Abstract
Intravenous Ig (IVIg) mediates protection from the effects of immune thrombocytopenic purpura (ITP) as well as numerous other autoimmune states; however, the active antibodies within IVIg are unknown. There is some evidence that antibodies specific for a cell-associated antigen on erythrocytes are responsible, at least in part, for the therapeutic effect of IVIg in ITP. Yet whether an IVIg directed to a soluble antigen can likewise be beneficial in ITP or other autoimmune diseases is also unknown. A murine model of ITP was used to determine the effectiveness of IgG specific to soluble antigens in treating immune thrombocytopenic purpura. Mice experimentally treated with soluble OVA + anti-OVA versus mice treated with OVA conjugated to rbcs (OVA-rbcs) + anti-OVA were compared. In both situations, mice were protected from ITP. Both these experimental therapeutic regimes acted in a complement-independent fashion and both also blocked reticuloendothelial function. In contrast to OVA-rbcs + anti-OVA, soluble OVA + anti-OVA (as well as IVIg) did not have any effect on thrombocytopenia in mice lacking the inhibitory receptor FcγRIIB (FcγRIIB–/– mice). Similarly, antibodies reactive with the endogenous soluble antigens albumin and transferrin also ameliorated ITP in an FcγRIIB-dependent manner. Finally, broadening the significance of these experiments was the finding that anti-albumin was protective in a K/BxN serum–induced arthritis model. We conclude that IgG antibodies directed to soluble antigens ameliorated 2 disparate IVIg-treatable autoimmune diseases.
Authors
Vinayakumar Siragam, Davor Brinc, Andrew R. Crow, Seng Song, John Freedman, Alan H. Lazarus
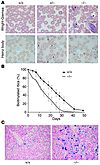
Abstract
Hemoglobin (Hb) A production during red blood cell development is coordinated to minimize the deleterious effects of free α- and β-Hb subunits, which are unstable and cytotoxic. The α-Hb–stabilizing protein (AHSP) is an erythroid protein that specifically binds α-Hb and prevents its precipitation in vitro, which suggests that it may function to limit free α-Hb toxicities in vivo. We investigated this possibility through gene ablation and biochemical studies. AHSP–/– erythrocytes contained hemoglobin precipitates and were short-lived. In hematopoietic tissues, erythroid precursors were elevated in number but exhibited increased apoptosis. Consistent with unstable α-Hb, AHSP–/– erythrocytes contained increased ROS and evidence of oxidative damage. Moreover, purified recombinant AHSP inhibited ROS production by α-Hb in solution. Finally, loss of AHSP worsened the phenotype of β-thalassemia, a common inherited anemia characterized by excess free α-Hb. Together, the data support a model in which AHSP binds α-Hb transiently to stabilize its conformation and render it biochemically inert prior to Hb A assembly. This function is essential for normal erythropoiesis and, to a greater extent, in β-thalassemia. Our findings raise the possibility that altered AHSP expression levels could modulate the severity of β-thalassemia in humans.
Authors
Yi Kong, Suiping Zhou, Anthony J. Kihm, Anne M. Katein, Xiang Yu, David A. Gell, Joel P. Mackay, Kazuhiko Adachi, Linda Foster-Brown, Calvert S. Louden, Andrew J. Gow, Mitchell J. Weiss

Abstract
Transplantation of genetically corrected autologous hematopoietic stem cells is an attractive approach for the cure of sickle-cell disease and β-thalassemia. Here, we infected human cord blood cells with a self-inactivating lentiviral vector encoding an anti-sickling βA-T87Q-globin transgene and analyzed the transduced progeny produced over a 6-month period after transplantation of the infected cells directly into sublethally irradiated NOD/LtSz-scid/scid mice. Approximately half of the human erythroid and myeloid progenitors regenerated in the mice containing the transgene, and erythroid cells derived in vitro from these in vivo–regenerated cells produced high levels of βA-T87Q-globin protein. Linker-mediated PCR analysis identified multiple transgene-positive clones in all mice analyzed with 2.1 ± 0.1 integrated proviral copies per cell. Genomic sequencing of vector-containing fragments showed that 86% of the proviral inserts had occurred within genes, including several genes implicated in human leukemia. These findings indicate effective transduction of very primitive human cord blood cells with a candidate therapeutic lentiviral vector resulting in the long-term and robust, erythroid-specific production of therapeutically relevant levels of β-globin protein. However, the frequency of proviral integration within genes that regulate hematopoiesis points to a need for additional safety modifications.
Authors
Suzan Imren, Mary E. Fabry, Karen A. Westerman, Robert Pawliuk, Patrick Tang, Patricia M. Rosten, Ronald L. Nagel, Philippe Leboulch, Connie J. Eaves, R. Keith Humphries

Abstract
Myelodysplasia is a hematological disease in which genomic abnormalities accumulate in a hematopoietic stem cell leading to severe pancytopenia, multilineage differentiation impairment, and bone marrow (BM) apoptosis. Mortality in the disease results from pancytopenia or transformation to acute myeloid leukemia. There are frequent cytogenetic abnormalities, including deletions of chromosomes 5, 7, or both. Recurring chromosomal translocations in myelodysplasia are rare, but the most frequent are the t(3;3)(q21;q26) and the inv(3)(q21q26), which lead to the inappropriate activation of the EVI1 gene located at 3q26. To better understand the role of EVI1 in this disease, we have generated a murine model of EVI1-positive myelodysplasia by BM infection and transplantation. We find that EVI1 induces a fatal disease of several stages that is characterized by severe pancytopenia. The disease does not progress to acute myeloid leukemia. Comparison of in vitro and in vivo results suggests that EVI1 acts at two levels. The immediate effects of EVI1 are hyperproliferation of BM cells and downregulation of EpoR and c-Mpl, which are important for terminal erythroid differentiation and platelet formation. These defects are not fatal, and the mice survive for about 10 months with compensated hematopoiesis. Over this time, compensation fails, and the mice succumb to fatal peripheral cytopenia.
Authors
Silvia Buonamici, Donglan Li, Yiqing Chi, Rui Zhao, Xuerong Wang, Larry Brace, Hongyu Ni, Yogen Saunthararajah, Giuseppina Nucifora







Copyright © 2024 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)